

Elucidating vasculitis
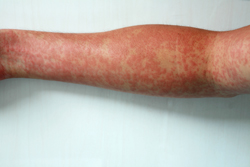
Vasculitis can cause undesirable changes like thickening, scarring, narrowing and weakening in the vessel walls. Inflammation is the result of the body's autoimmune system attacking the blood vessels and the aetiology is unclear. Autoimmune vasculitis has been associated with the activation of neutrophils; regulating neutrophil apoptosis (cell death) could reduce inflammation to ameliorate vasculitis symptoms. The A20 (zinc-finger) protein produces anti-inflammatory activity by inhibiting activation of nuclear factor kappa-light-chain-enhancer of B (NF-kB) cells. NF-kB cells are activated in response to tumour necrosis factor (TNF)-alpha and lipopolysaccharide (LPS)-mediated NF-kB pathways (associated with endothelial and neutrophil cell survival). The EU-funded A20APOPTOSIS project will assess the structure and function of the A20 protein in regulating neutrophil and endothelial cell apoptosis. Elucidating the role of A20 and A20-binding proteins will enable the development of novel therapeutic targets for vasculitis. Molecular biology, cell biology, functional biology, molecular modelling and clinical investigations will be used for this purpose. Research results revealed the presence of the A20 protein in inflammatory neutrophils. Work is ongoing to identify if A20 has a pro- or anti-apoptotic role in neutrophils. Project outcomes could uncover an as yet unexplored role of A20 expression in modulating neutrophil apoptosis and thereby a novel means to treat vasculitis.



