Protein degradation in neurodegeneration
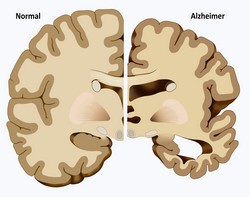
Protein aggregation is a hallmark of many neurodegenerative disorders including Alzheimer's disease, Parkinson's disease and polyglutamine diseases (polyQ). To date, there is no causal treatment available for polyQ disorders, necessitating the urgent development of novel therapeutic strategies. The EU-funded TREATPOLYQ(opens in new window) (TreatPolyQ – Industrial academic initial training network towards treatment of polyglutamine diseases) network set out to investigate several important disease-associated mechanisms and translate them into promising treatment strategies. The work focused on the two most common polyQ diseases, Huntington's disease (HD) and spinocerebellar ataxia type 3 (SCA3). Scientists observed that the nuclear localisation of the deubiquitinating enzyme ataxin 3 was of critical importance for the pathogenesis of polyQ diseases and a promising target for pharmaceutical interventions. Transport proteins involved in the formation of the intra-nuclear aggregates could be modulated to alleviate the phenotype of ataxin 3 mutations. Under physiological conditions, huntingtin (HTT) influences the vesicular transport of neurotrophic factors via phosphorylation but in HD, this function is disturbed and leads to neuronal death. TREATPOLYQ researchers investigated the role of HTT protein by focusing on its conformations and interaction with protein complexes. To comprehend the mechanism of ubiquitination in neurodegenerative disorders further, researchers studied the mechanisms by which polyQ-containing proteins are degraded. Since macroautophagy is the only pathway that can degrade protein aggregates, they studied this process in vitro to discover peptides capable of interfering with HTT fibril formation. Their findings clearly indicated that the deficits in proteasomal degradation associated with HD pathology might be prevented by a cell-specific upregulation of autophagy. From a diagnostics perspective, the research teams developed novel assays for detecting the different HTT forms and identified enzymes responsible for maintaining the correct labelling of HTT and ataxin 3 for degradation. Although further proof for the disease-modifying capacity of these enzymes is required, enzyme inhibitors could serve as a potential treatment for HD and SCA3. Overall, the TREATPOLYQ consortium succeeded in deciphering important mechanisms associated with polyQ diseases and advanced our understanding of disease aetiology. From a therapeutic angle, project findings will undoubtedly impact the clinical outcome of HD and SCA3 patients in the future through novel treatment approaches.