

De nouvelles techniques de pointe pour la simulation de molécules
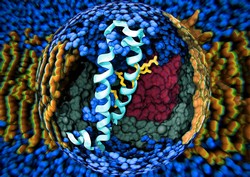
Financé par l'UE, le projet ERIKLINDAHLERC2007 (Multiscale and distributed computing algorithms for biomolecular simulation and efficient free energy calculations) a fait progresser les simulations de biomolécules. Il a augmenté la performance de ces outils de plusieurs ordres de magnitude, et renforcé l'exactitude des prévisions. Les chercheurs ont accompli des progrès remarquables, à commencer par le développement de la version 4.6 de GROMACS(s’ouvre dans une nouvelle fenêtre). Ce logiciel peut traiter différentes sortes de biomolécules (par ex, les protéines, les lipides et les acides nucléiques) parallèlement à leurs champs de force correspondants via des caractéristiques spécifiques de CHARMM27. Les chercheurs ont amélioré les performances maximales d'environ 40 fois, et de dix fois pour le calcul de simulations en parallèle. Par ailleurs, ce code de simulation en accès libre était cinq fois plus rapide que l'état actuel de l'art. L'implémentation de calculs d'énergie libre sur la base du «Bennett Acceptance Ratio» a permis une analyse rapide et plus exacte même dans des environnements informatiques distribués et sur le Cloud. Par ailleurs, les membres du projet ont automatisé la création de données entrantes de l'énergie libre alors que les analyses statistiques ont fourni des estimations d'écart-type pour tous les calculs de l'énergie libre. Le projet a cependant culminé par le développement d'un nouveau cadre permettant une «dynamique moléculaire adaptative parallèle», qui combine l'informatique distribuée aux simulations de dynamique des molécules. Disponible sur le site web Copernicus(s’ouvre dans une nouvelle fenêtre), cet outil en open source planifie automatiquement des milliers de simulations étroitement couplées tandis que l'informatique distribuée augmente de quelques ordres de magnitude l'efficacité de l'échantillonnage. Le nombre de simulations requises a été réduit grâce à des simulations adaptatives pour le calcul de l'énergie libre et à la génération automatisée des paramètres de simulation pour de petites molécules. Par conséquent, Copernicus peut déterminer en quelques heures les énergies libres de solvatation et de liaison pour une longue liste de composés. Et ce, pour moins d'un euro par composé. Ces nouveaux outils ont aidé les chercheurs à mener des simulations d'importants systèmes biologiques comme les canaux ioniques contrôlés et les protéines membranaires. Les découvertes clés ont conduit à de nombreuses publications dans des revues avec des implications significatives pour la conception de médicaments. Par exemple, l'étude des canaux ioniques contrôlés par des ligands a fourni de nouvelles informations sur le mécanisme de modulation allostérique à double site. Cela pourrait s'avérer utile dans la conception de nouvelles paires de médicaments anesthésiques on/off. Dans l'ensemble, les outils et les résultats découlant de l'étude ERIKLINDAHLERC2007 pourraient enrichir nos connaissances sur les maladies dégénératives comme celle de Creutzfeldt-Jakob, et contribuer à la conception rationnelle de médicaments. Leur application pourrait également s'étendre au-delà de la biomédecine, à la science des polymères et aux nanotechnologies.



